
- García-Pereira, MJ., Quesada,H., Caballero, A. and Carvajal-Rodríguez,A. AFLPMax: a user-friendly application for computing the optimal number of AFLP markers needed in phylogenetic reconstruction. Molecular Ecology Resources (in Press).
If you use AFLPMax in a publication you should also acknowledge the use of Seq-Gen, PHYLIP and Ktreedist software in addition to ours. So, please cite the following references:
- Felsenstein, J. (2005). PHYLIP (Phylogeny Inference Package) version 3.6. Distributed by the author. Department of Genome Sciences, University of Washington, Seattle.
- Rambaut, A. and Grassly, N. C. (1997). Seq-Gen: An application for the Monte Carlo simulation of DNA sequence evolution along phylogenetic trees. Comput. Appl. Biosci. 13: 235-238.
- Soria-Carrasco, V., Talavera, G., Igea, J. and Castresana, J. (2007). The K tree score: quantification of differences in the relative branch length and topology of phylogenetic trees. Bioinformatics 23, 2954-2956.
Ktreedist:
Institute of Molecular Biology of Barcelona (IBMB), CSIC, Jordi Girona 18, 08034 Barcelona, Spain vscagr@ibmb.csic.es (VSC) & jcvagr@ibmb.csic.es (JC)
This program is free software; you can redistribute it and/or modify it under the terms of the GNU General Public License as published by the Free Software Foundation; either version 2 of the License, or (at your option) any later version. This program is distributed in the hope that it will be useful, but WITHOUT ANY WARRANTY; without even the implied warranty of MERCHANTABILITY or FITNESS FOR A PARTICULAR PURPOSE. See the GNU General Public License for more details. You should have received a copy of the GNU General Public License along with this program; if not, write to the Free Software Foundation, Inc., 51 Franklin Street, Fifth Floor, Boston, MA 02110-1301, USA.
The following copyright notice is intended to cover all source code, all documentation, and all executable programs of the PHYLIP package.
© Copyright 1980-2008. University of Washington. All
rights reserved. Permission is granted to reproduce, perform, and modify
these programs and documentation files. Permission is granted to distribute
or provide access to these
programs provided that this copyright notice is not removed, the programs are
not integrated with or called by any product or service that generates
revenue, and that your distribution of these documentation files and programs
are free. Any modified
versions of these materials that are distributed or accessible shall indicate
that they are based on these programs. Institutions of higher education are
granted permission to distribute this material to their students and staff
for a fee to recover distribution costs. Permission requests for any other
distribution of these program should be directed to:
license (at) u.washington.edu .
Seq-Gen:
Copyright (c)1996-2005, Andrew Rambaut & Nick Grassly
Department of Zoology, University of Oxford
All rights reserved.
Redistribution and use in source and binary forms, with or without modification, are permitted provided that the following conditions are met:
1. Redistributions of source code must retain the above copyright notice, this list of conditions and the following disclaimer.
2. Redistributions in binary form must reproduce the above copyright notice, this list of conditions and the following disclaimer in the documentation and/or other materials provided with the distribution.
3. The names of its contributors may not be used to endorse or promote products derived from this software without specific prior written permission.
THIS SOFTWARE IS PROVIDED BY THE COPYRIGHT HOLDERS AND CONTRIBUTORS "AS IS" AND ANY EXPRESS OR IMPLIED WARRANTIES, INCLUDING, BUT NOT LIMITED TO, THE IMPLIED WARRANTIES OF MERCHANTABILITY AND FITNESS FOR A PARTICULAR PURPOSE ARE DISCLAIMED. IN NO EVENT SHALL THE COPYRIGHT OWNER OR CONTRIBUTORS BE LIABLE FOR ANY DIRECT, INDIRECT, INCIDENTAL, SPECIAL, EXEMPLARY, OR CONSEQUENTIAL DAMAGES (INCLUDING, BUT NOT LIMITED TO, PROCUREMENT OF SUBSTITUTE GOODS OR SERVICES; LOSS OF USE, DATA, OR PROFITS; OR BUSINESS INTERRUPTION) HOWEVER CAUSED AND ON ANY THEORY OF LIABILITY, WHETHER IN CONTRACT, STRICT LIABILITY, OR TORT (INCLUDING NEGLIGENCE OR OTHERWISE) ARISING IN ANY WAY OUT OF THE USE OF THIS SOFTWARE, EVEN IF ADVISED OF THE POSSIBILITY OF SUCH DAMAGE.

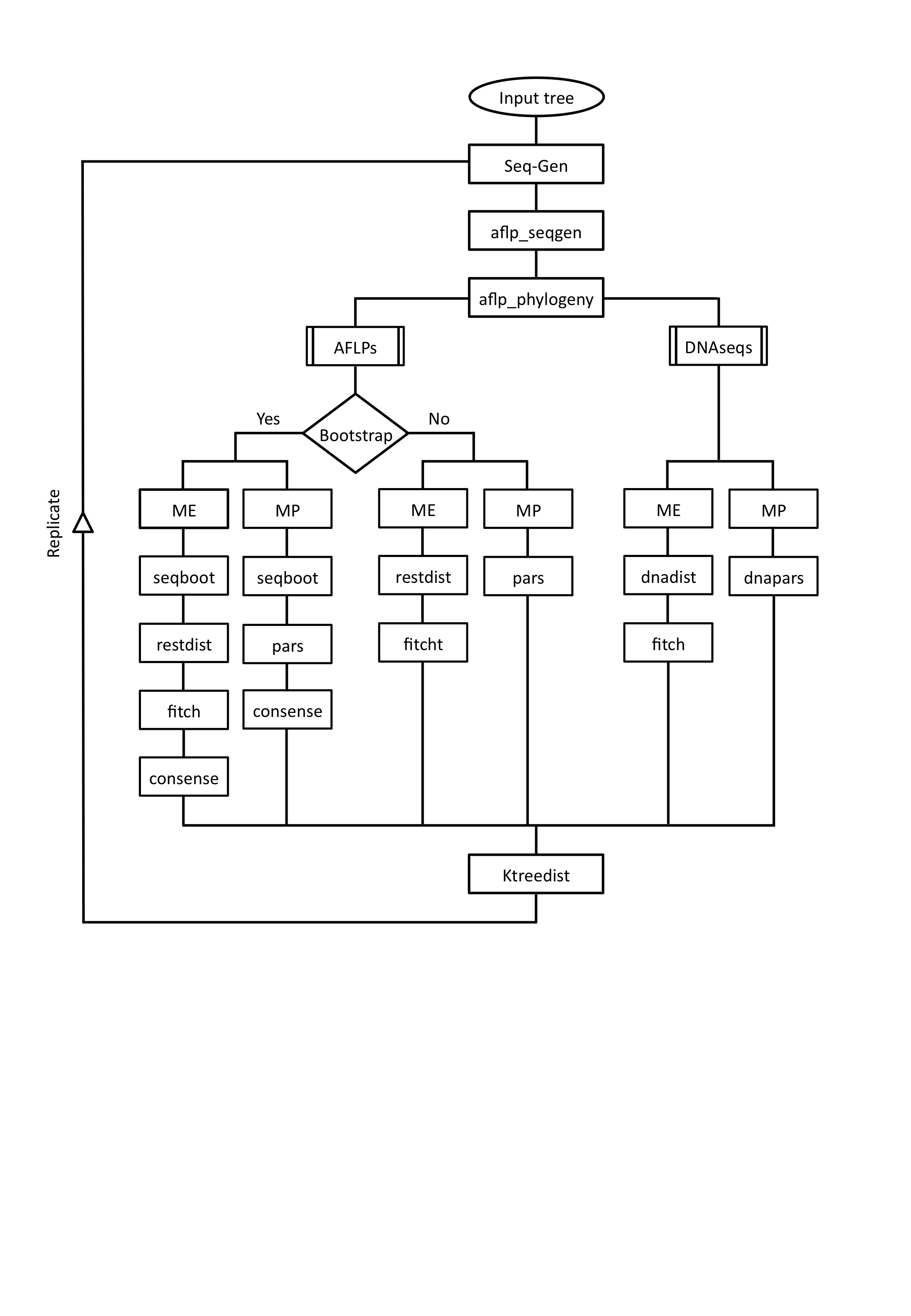
In brief, AFLPMax is a combination of programs that, using a tree defined by the user, performs the following tasks:
Taxa names cannot start with a number but any alphanumeric character is admitted in any other position of the name. The first and only step needed to run the program is to load a tree. You can paste it directly into the INPUT TREE tab or you can load it from the File menu, option Open, also with the shortcut Ctrl+O. Once you have loaded a tree the Run button is activated and you can run an analysis using the default options or you can change them in the settings button and menu.
The reference tree defined by the user (see Input file section) is used to obtain DNA sequences with the program Seg-Gen (Rambaut & Grassly, 1997). Seq-Gen evolves sequences along a specific tree under a given evolution model (chosen by the user).
A computer program (aflp_seqgen) written in C is then used to simulate the AFLP technique. Each sequence corresponding to each taxon is cut separately with enzymes EcoRI and MseI, which are the typical enzymes used in AFLP studies. The program searches for all the restriction sites and returns all fragments that would result from the digestion. The output is the list of fragments sorted by their length, as it would appear in a real experiment. The in silico AFLP profiles are generated using the 4096 possible combination of three selective nucleotides for each enzyme. Another program (aflp_phylogeny), also written in C, is used to combine the information of all individuals and to construct the 1/0 presence/absence matrices. The program selects randomly the different number of bands used to reconstruct the phylogenies, starting from 100 and increasing by 100 each time (each set of 100 bands would correspond to a different primer combination) until a maximum of 1000 bands. Those numbers are chosen because 100 bands per AFLP profile are recommended for experimental data sets.
AFLP-based phylogenies are estimated with programs from the PHYLIP package (Felsenstein, 2005), using two of the most widely used methods in AFLP data sets: minimum evolution (ME) and maximum parsimony (MP). For the distance-based method (ME), each binary file of presence/absence is converted into a distance matrix with the program restdist using the Nei and Li distance (1979). The distance matrix obtained is used as input for the program fitch to infer the phylogeny under the ME algorithm. For the character based method (MP) the presence/absence matrix is used directly as input for the program pars. The user is allowed to generate the desired number of bootstrap pseudo-samples from the original data. To do that the program seqboot from the PHYLIP package is used. The obtained matrices are used to reconstruct the phylogenies with the same programs as before. A consense tree for MP and other for ME are obtained with the program consense with the minimum cut-off value indicated by the user. The phylogenetic inference can also be done with DNA sequences, which means that sequences of 10000 nucleotides are simulated with Seq-Gen and used directly to infer the tree. In this case the program dnadist is used to compute the distance matrix needed to reconstruct the phylogeny under the ME method with the program fitch. To obtain the MP tree the program dnapars is used.
Each estimated tree is then compared with the reference tree using the program Ktreedist (Soria-Carrasco et al., 2007), which takes into account both topology and branch length information of a phylogenetic tree. This program computes a K-score that measures overall differences in the relative branch length and topology of two phylogenetic trees after scaling one of the trees to have a global divergence as similar as possible to the other tree. The program also computes the symmetric difference or Robinson-Foulds (R-F) distance (Robinson and Foulds, 1981), which only takes into account the topology of the phylogenetic trees.
AFLPMax generates a table for each analysis and inference method showing the mean and standard error values obtained for the K-score and the R-F distance.
The outputs are two tables for each analysis, one corresponding to the ME results and another corresponding to the MP ones. Those tables are displayed in the corresponding tabs of the interface but are also saved in your computer as html files in a folder under your election. The names of the files start with ME_ or MP_ indicating the phylogenetic method used, the next part of the name refers to the analysis done (BAND: AFLP bands, BOOT: AFLP bands with bootstrapping or SEQ: DNA sequences). The last part of the name, RES, is common to the three possible analyses and is just to indicate that the file contains the table of results. For example, a file called ME_BAND_RES.html would contains the results of the AFLP analysis reconstructing the phylogenies with the ME inference method. Below you can find some examples of the tables of results, depending on the analysis done. If you run AFLPMax selecting AFLP analysis the output will show the number of bands used to reconstruct the phylogeny on the first column and the mean and standard error obtained from the replicates for the K-score and the R-F distance on the following ones. Values of K-score and R-F distance equal to zero indicate perfect match between the reference tree and the AFLP-reconstructed tree.
MP_BAND_RES:
| num band | mean K | se K | mean RF | se RF |
|---|---|---|---|---|
| 100 | 0.005794 | 0.000518 | 1.400000 | 0.221108 |
| 200 | 0.005521 | 0.000611 | 1.300000 | 0.260342 |
| 300 | 0.005034 | 0.000561 | 0.400000 | 0.221108 |
| 400 | 0.004514 | 0.000492 | 0.400000 | 0.266667 |
| 500 | 0.004157 | 0.000365 | 0.200000 | 0.200000 |
| 600 | 0.004545 | 0.000411 | 0.100000 | 0.100000 |
| 700 | 0.004402 | 0.000379 | 0.000000 | 0.000000 |
| 800 | 0.004186 | 0.000247 | 0.000000 | 0.000000 |
| 900 | 0.004398 | 0.000294 | 0.000000 | 0.000000 |
| 1000 | 0.004269 | 0.000356 | 0.000000 | 0.000000 |
ME_BAND_RES:
| num band | mean K | se K | mean RF | se RF |
|---|---|---|---|---|
| 100 | 0.005059 | 0.000611 | 2.400000 | 0.498888 |
| 200 | 0.004461 | 0.000487 | 1.000000 | 0.447214 |
| 300 | 0.003799 | 0.000451 | 0.200000 | 0.200000 |
| 400 | 0.002553 | 0.000174 | 0.600000 | 0.305505 |
| 500 | 0.002424 | 0.000235 | 0.600000 | 0.305505 |
| 600 | 0.002713 | 0.000345 | 0.000000 | 0.000000 |
| 700 | 0.002361 | 0.000337 | 0.000000 | 0.000000 |
| 800 | 0.002071 | 0.000205 | 0.000000 | 0.000000 |
| 900 | 0.001942 | 0.000224 | 0.000000 | 0.000000 |
| 1000 | 0.002322 | 0.000320 | 0.200000 | 0.200000 |